Next: Open-source Production MD: Gromacs Up: All-atom Potential Energy Functions Previous: ReaxFF
As a precursor to the Tersoff formalism, perhaps the earliest attempt to
use molecular simulation with a reactive potential to study a realistic atomic-scale model of
silicon was due to Stillinger and Weber [38].
The C-code that implements the Stillinger-Weber (SW) potential
for silicon is mdswsi.c. This
code computes in reduced units as well; ![]() = 0.20951 nm,
= 0.20951 nm,
![]() = 2.1678 eV, and
= 2.1678 eV, and ![]() = 28.085 amu, which are appropriate
for a system of pure silicon. One reduced unit of temperature,
= 28.085 amu, which are appropriate
for a system of pure silicon. One reduced unit of temperature,
![]() , corresponds to 25156.74 K.
, corresponds to 25156.74 K.
The main reason to introduce Stillinger-Weber silicon here is to give you a historical example of an implementation of a simple reactive force-field. Silicon forms 4-coordinated tetrahedral bonded structures. The SW potential includes three-body interactions that enforce this symmetry as well as permit breaking and reforming of bonds to produce defects, for example.
The total potential is expressed as two sums, one for unique pair interactions, and another for unique triplet interactions:
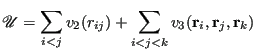 |
(282) |
![$\displaystyle v_2 \left(r\right) = \left\{\begin{array}{ll} \epsilon A \left(Br...
...\right)^{-1}\right], & \mbox{$r < a$} 0 & \mbox{$r\ge a$} \end{array} \right.$](img766.png) |
(283) |
The three body models the angles, and is the sum of functions of each
of the three angles of a triplet, ![]() :
:
| (284) |
Here I have employed the shorthand notation
![]() . Note that, in the
notation of this potential,
. Note that, in the
notation of this potential,
![]() is subtended at
is subtended at ![]() , and
, and
![]() :
:
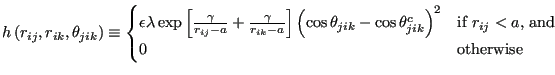 |
(285) |
One computes the angle-![]() term,
term, ![]() , and the angle-
, and the angle-![]() term,
term, ![]() , by permuting the indices appropriately.
, by permuting the indices appropriately.
The parameters used in the original study by Stillinger and Weber are:
| (286) | |||
| (287) | |||
| (288) | |||
| 0 | (289) | ||
| (290) | |||
| (291) | |||
| (292) |
As in any MD simulation, one computes the force on any particle ![]() from the negative gradient of the total potential:
from the negative gradient of the total potential:
| (293) | |||
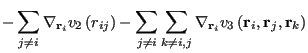 |
(294) |
The two-body term for the ![]() -
-![]() interaction is only slightly more
complicated than Lennard-Jones, due to the smooth cutoff. Here,
assuming
interaction is only slightly more
complicated than Lennard-Jones, due to the smooth cutoff. Here,
assuming ![]() and
and ![]() are within interaction range (
are within interaction range (
![]() ), we
have for the force on
), we
have for the force on ![]() due to
due to ![]() :
:
It is comparatively much more tedious to evaluate the three-body gradients:
Each of the partials in Eq. 303 is unique:
Let us now run two MD simulations for 10,000 time steps; one with and
and the other without three body forces, at reduced initial
temperature ![]() = 0.12 and reduced density
= 0.12 and reduced density ![]() = 0.46 for a small
system of 216 atoms. The code
= 0.46 for a small
system of 216 atoms. The code mdswsi.c can initialize atoms on a diamond-cubic lattice; the DC unit cell has 8 atoms, so the number of atoms one should specify for a perfect crystal is 8 times any product of three integers. Using the -nc #,#,# switch we indicate how may DC unit cells we want in the ![]() ,
, ![]() , and
, and ![]() , directions, respectively. The code also has a switch
, directions, respectively. The code also has a switch -do-three-body which is by default on, but can be turned off by passing a zero. Running this twice generates two logs and two trajectories:
$ ./mdswsi -nc 3,3,3 -ns 10000 -fs 10 -prog 10 \ -do-three-body 1 -traj yes3.xyz > yes3.log $ ./mdswsi -nc 3,3,3 -ns 10000 -fs 10 -prog 10 \ -do-three-body 0 -traj no3.xyz > no3.log
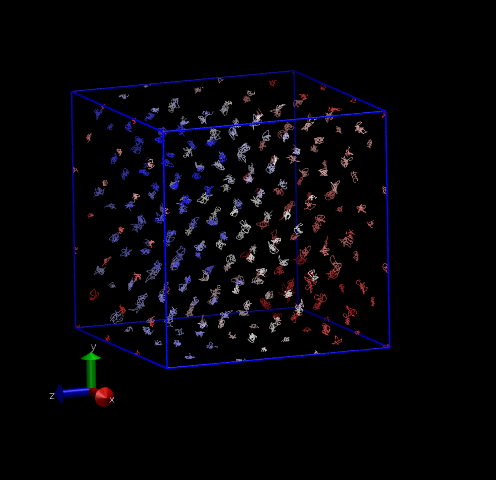
Fig. 30 shows two 3-D system representations. The first is a perfect DC lattice with 216 atoms, where a dot is drawn every 10 time steps for each atom, and each atom is colored uniquely. The second shows the same view of a system for which the three-body forces are turned off; notice that it appears liquid-like. Three-body forces are required to stabilize the DC lattice since each atom only has four nearest neighbors.
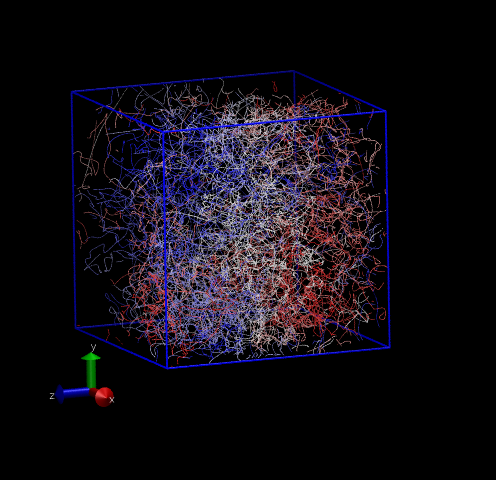 |
Fig. 31 shows a plot of two-body and three-body potential energy vs. time from the first simulation, and a plot of instantaneous temperature vs. time from both simulations on the right. Notice that the three-body forces act to keep the system oscillating in its crystalline state, and the lack of three-body forces results in system melting. This latter occurrence is because the DC lattice is a fairly unfavorable configuration for a system with only two-body forces active.
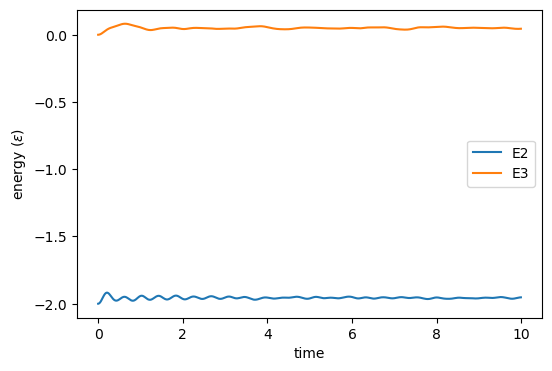
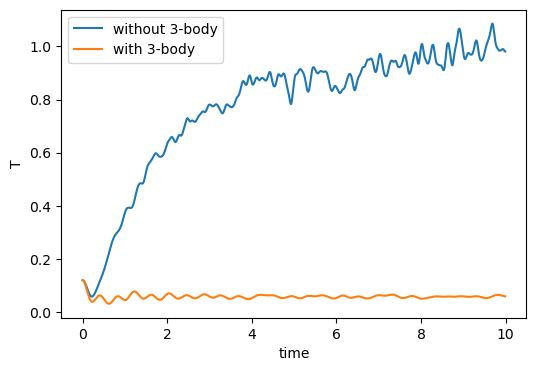 |
The code mdswsi.c is very slow when three-body forces are turned on because it uses a brute-force ![]() loop. In practice, any code that implements two- and three-body potentials with short-range cutoffs will use data structures like the neighbor list and the link-cell algorithm to make the pair and triplet loops more efficient. And although SW silicon is historically important, there are much better (and more complicated) potentials out there for silicon-based systems, such as COMB [34].
loop. In practice, any code that implements two- and three-body potentials with short-range cutoffs will use data structures like the neighbor list and the link-cell algorithm to make the pair and triplet loops more efficient. And although SW silicon is historically important, there are much better (and more complicated) potentials out there for silicon-based systems, such as COMB [34].
![$\displaystyle -\frac{\partial }{\partial{\bf r}_i} \left\{\epsilon A \left(Br_{ij}^{-p}-r_{ij}^{-q}\right)\exp\left[\left(r_{ij}-a\right)^{-1}\right]\right\}$](img794.png)
![$\displaystyle -\epsilon A \frac{{\bf r}_{ij}}{r_{ij}}\left(\left.\left[\frac{\p...
...)\right]\right\vert _{r_{ij}}\exp\left[\left(r_{ij}-a\right)^{-1}\right]\right.$](img795.png)
![$\displaystyle \left.\left(Br_{ij}^{-p}-r_{ij}^{-q}\right)\left.\left\{\frac{\pa...
...al r}\exp\left[\left(r-a\right)^{-1}\right]\right\}\right\vert _{r_{ij}}\right)$](img797.png)
![$\displaystyle v_2\frac{{\bf r}_{ij}}{r_{ij}}\left[\frac{pBr_{ij}^{-p-1}-qr_{ij}^{-q-1}}{Br_{ij}^{-p}-r_{ij}^{-q}} + \left(r_{ij}-a\right)^{-2}\right]$](img801.png)
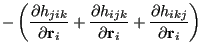
![\begin{displaymath}\begin{array}{lll} \mbox{\raisebox{-0.5cm}{\includegraphics[w...
...ik}}\frac{1}{r_{ij}}\right) \cos\theta_{jik}\right] \end{array}\end{displaymath}](img811.png)
![\begin{displaymath}\begin{array}{lll} \mbox{\raisebox{-0.5cm}{\includegraphics[w...
...j}}{r_{ij}}\frac{1}{r_{ij}} \cos\theta_{ijk}\right] \end{array}\end{displaymath}](img812.png)
![\begin{displaymath}\begin{array}{lll} \mbox{\raisebox{-0.5cm}{\includegraphics[w...
...k}}{r_{ik}}\frac{1}{r_{ik}} \cos\theta_{ikj}\right] \end{array}\end{displaymath}](img813.png)