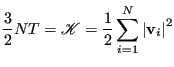Next: The Code Up: Case Study 1: An Previous: Case Study 1: An
Let us consider a few of the important elements of any MD program:
We will understand these elements by manipulating an existing
simulation program that implements the Lennard-Jones fluid
(which you may recall was analyzed using Metropolis Monte-Carlo
simulation in Sec. 3). The program is called mdlj.c in the
instructional codes repository.
Recall the Lennard-Jones pair potential:
| (151) |
We have already encountered interparticle forces for the Lennard-Jones
pair potential in the context of computing the pressure from the
virial in the MC simulation of the LJ fluid (Sec. 3.6). Briefly,
the force exerted on particle ![]() by virtue of its Lennard-Jones interaction
with particle
by virtue of its Lennard-Jones interaction
with particle ![]() ,
,
![]() , is given by:
, is given by:
![$\displaystyle {\bf f}_{ij}\left(r_{ij}\right) = \frac{{\bf r}_{ij}}{r_{ij}^2}\l...
...{1}{2}\left(\frac{\sigma}{r_{ij}}\right)^6\right]\right\} \equiv {\bf r}_{ij}f.$](img454.png) |
(153) |
Below is a C-code fragment for computing both the total potential energy and interparticle forces:
double forces ( double * rx, double * ry, double * rz,
double * fx, double * fy, double * fz,
int n ) {
int i,j;
double dx, dy, dz, r2, r6, r12;
double e = 0.0, f = 0.0;
for (i=0;i<n;i++) {
fx[i] = 0.0;
fy[i] = 0.0;
fz[i] = 0.0;
}
for (i=0;i<(n-1);i++) {
for (j=i+1;j<n;j++) {
dx = rx[i]-rx[j];
dy = ry[i]-ry[j];
dz = rz[i]-rz[j];
r2 = dx*dx + dy*dy + dz*dz;
r6i = 1.0/(r2*r2*r2);
r12i = r6i*r6i;
e += 4*(r12i - r6i);
f = 48/r2*(r6i*r6i-0.5*r6i);
fx[i] += dx*f;
fx[j] -= dx*f;
fy[i] += dy*f;
fy[j] -= dy*f;
fz[i] += dz*f;
fz[j] -= dz*f;
}
}
return e;
}
Notice that the argument list now includes arrays for the forces, and
because force is a vector quantity, we have three parallel arrays for
a three-dimensional system. These forces must of course be
initialized, shown in lines 6-10. The
The second major aspect of MD is the integrator. As discussed in
class, we will primarily use Verlet-style (explicit) integrators. The
most common version is the velocity-Verlet algorithm [5],
first presented in Sec. 4.1.1. Below is a fragment of C-code
for executing one time step of integration for a system of ![]() particles:
particles:
for (i=0;i<N;i++) {
rx[i]+=vx[i]*dt+0.5*dt2*fx[i];
ry[i]+=vy[i]*dt+0.5*dt2*fy[i];
rz[i]+=vz[i]*dt+0.5*dt2*fz[i];
vx[i]+=0.5*dt*fx[i];
vy[i]+=0.5*dt*fy[i];
vz[i]+=0.5*dt*fz[i];
}
PE = total_e(rx,ry,rz,fx,fy,fz,N,L,rc2,ecor,ecut,&vir);
KE = 0.0;
for (i=0;i<N;i++) {
vx[i]+=0.5*dt*fx[i];
vy[i]+=0.5*dt*fy[i];
vz[i]+=0.5*dt*fz[i];
KE+=vx[i]*vx[i]+vy[i]*vy[i]+vz[i]*vz[i];
}
KE*=0.5;
Notice the update of positions
(Eq. 125), where vx[i] is the ![]() -component of
velocity,
-component of
velocity, fx[i] is the ![]() -component of force,
-component of force, dt and dt2 are the time-step and squared time-step, respectively. Notice
that there is no implementation of periodic boundaries in this
code fragment; what would this “missing code” look like? (Hint: see
mdlj.c for the answer!) Lines 5-7 are the first half-update of
velocities (Eq. 126). The force routine computes
the new forces on the currently updated configuration on line 10.
Then, lines 12-18 perform the second-half of the velocity update
(Eq. 127). Also note that the kinetic energy,
![]() , is computed in this loop.
, is computed in this loop.
cfa22@drexel.edu
![$\displaystyle u\left(r\right) = 4\epsilon\left[\left(\frac{\sigma}{r}\right)^{12}- \left(\frac{\sigma}{r}\right)^{6}\right]$](img450.png)