Next: Free Energy Methods Up: Open-source Production MD: Gromacs Previous: The SARS-CoV-2 spike protein
The NAMD source code and many precompiled binaries are available from the official download site at the University of Illinois. Like Gromacs, NAMD is also available in many supercomputing centers. Compiling NAMD from source is not for the faint-hearted, so I won't cover that here; fortunately for us, the standard `Linux-multicore` executable will work just fine in Ubuntu on WSL. (If you have a mac, you'll have to download a mac-specific executable.) For now, just download the tarball, extract it, and copy the namd2 executable to /usr/local/bin/:
$ tar zxf NAMD_2.14_Linux-x86_64-multicore.tar.gz $ cd NAMD_2.14_Linux-x86_64-multicore/ $ sudo cp namd2 /usr/local/bin/
If you have not done so already, download the Linux version of VMD and install it inside WSL as well - we need to use that rather than the native Windows version here.
To build a system to simulation with NAMD with the CHARMM force field, one must use the psfgen utility of VMD. This will generate an input PDB file along with a PSF file that contains all the topological information. I cannot stress enough how important it is to use the psfgen manual; I'm only showing a little bit of here.
Let's revisit the SARS-CoV-2 S RBD for this example. Starting with the same coordinates extracted from the PDB entry and put into my_7c8j.pdb, we use a series of VMD scripts to build a solvated system.
First is mkpsf.tcl (it's name is not important; what's important is what is inside it). This script contains TcL commands that implement PSF generation using VMD as an interpreter. To run it:
$ vmd -dispdev text -e mkpsf.tcl
This generates my_7c8j.psf which is an X-PLOR Protein Sturcture Format (PSF) file, along with the PDB file my_78cj_processed.pdb, which contains all the missing H's. It is customary to run a short energy minimization in vacuum just in case some of those H's are a little too close to each other by accident. As with Gromacs, this is done by invoking the main MD program with an input configuration file requesting a minimization. Here, this is vac.namd. This can be run:
$ namd2 vac.namd >& vac.log & $ tail -f vac.log
The tail command allows you to watch the simulation write to vac.log as it runs. Once this finishes, the next step is to solvate and neutralize. This is accomplished with another script, solv.tcl, which uses both the solvate and autoionize VMD packages:
$ vmd -dispdev text -e solv.tcl
Similar to the editconf in Gromacs, solv.tcl computes and saves the resulting box dimensions, here in a file called cell.inp. Once this is done, we see the new PSF/PDB pair my_7c8j_i.psf and my_7c8j_i.pdb that contain the protein and all the solvent. These files, along with cell.inp and the CHARMM parameter files are now inputs for a 100-ps NPT MD simulation described by prod.namd.
$ namd2 +p8 prod.namd >& prod.log & $ tail -f prod.log
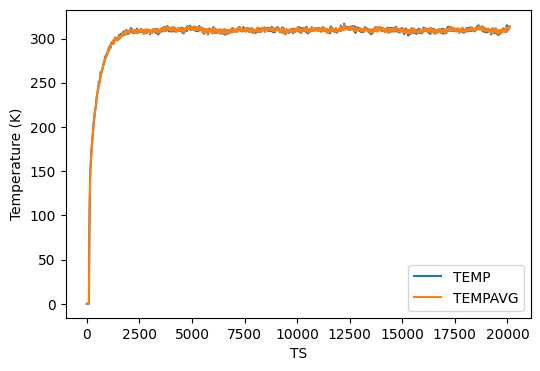
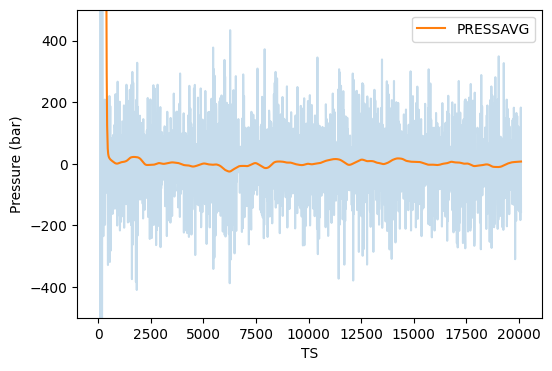
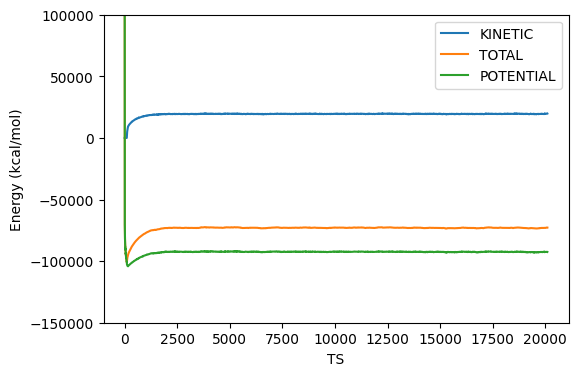
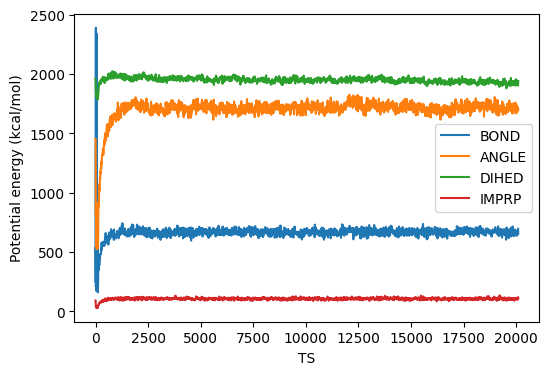
This takes about 1.5 h on my Dell Latitude 7400 2-in-1 Intel Core-I7 laptop on WSL2 running Ubuntu. I ran this in a directory called instructional-codes/my_work/namd, so the directory originals is two up from the run directory. Fig. 38 shows plots of energies, temperature, and pressure, vs time-step from this MD simulation, generated by the following four python commands:
$ python3 ../../originals/plot_mdlj_log.py -fmt NAMD \ -logs prod.log -d 11 14 -ylabel "Temperature (K)" -o temp.png $ python3 ../../originals/plot_mdlj_log.py -fmt NAMD \ -logs prod.log -d 18 -ylabel "Pressure (bar)" -o pres.png \ -ylim -500 500 -every 100 $ python3 ../../originals/plot_mdlj_log.py -fmt NAMD \ -logs prod.log -d 9 10 12 -ylabel "Energy (kcal/mol)" \ -ylim -150000 100000 -o kin-pot-tot.png $ python3 ../../originals/plot_mdlj_log.py -fmt NAMD \ -logs prod.log -d 1 2 3 4 -ylabel "Potential energy (kcal/mol)" \ -o bonded.png
 |
It should be stressed that system generation in both Gromacs and NAMD requires surmounting a significant learning curve if one wants to venture from only the simplest systems shown here. The Gromacs workflow is (to me) a bit more uniform than NAMD, which requires essentially a bunch of script-writing. (VMD offers an automated PSF builder that also works for simple systems.)
{kind=link}